

HADDOCK tutorial for the use of MS crosslink data to guide docking
This tutorial consists of the following sections:
- Introduction
- Setup/Requirements
- HADDOCK general concepts
- The information at hand
- Scenarios for docking - how to make use of MS data in HADDOCK
- Register for the HADDOCK web server
- Setting up the docking from cross-link restraints (scenario 1)
- Setting up the docking with the DisVis-derived interfaces (scenario 2)
- Setting up the docking with cross-links and the DisVis-derived interfaces (scenario 3)
- First analysis of the results
- Visualisation of docked models
- Comparison with the crystal structure of an homologous complex
- Conclusions
- Congratulations!
Introduction
This tutorial will demonstrate the use of HADDOCK for predicting the structure of a protein-protein complex from MS cross-linking data. The case we will be investigating is the interaction between two proteins of the 26S proteasome of S. pombe, PRE5 (UniProtKB: O14250) and PUP2 (UniProtKB: Q9UT97). For this complex seven experimentally determined cross-links (4 Adipic acid dihydrazide (ADH) & 3 Zero-length (ZL)) are available (Leitner et al., 2014). The tutorial builds on our DisVis tutorial to evaluate the information content of MS cross-links and identify possible false positive. Another feature of DisVis is, that it allows to identify the surface residues that are most often contacted in all possible models of the complex satisfying the cross-links. This is an additional information which might be useful to guide the docking.
We will thus be making use of the results of the DisVis tutorial to setup various docking runs using our HADDOCK2.4 webserver.
A description of our web server can be found in the following publications:
-
R.V. Honorato, M.E. Trellet, B. Jiménez-García, J.J. Schaarschmidt, M. Giulini, V. Reys, P.I. Koukos, J.P.G.L.M. Rodrigues, E. Karaca, G.C.P. van Zundert, J. Roel-Touris, C.W. van Noort, Z. Jandová, A.S.J. Melquiond and A.M.J.J. Bonvin. The HADDOCK2.4 web server: A leap forward in integrative modelling of biomolecular complexes. Nature Prot., Advanced Online Publication DOI: 10.1038/s41596-024-01011-0 (2024).
-
G.C.P van Zundert, J.P.G.L.M. Rodrigues, M. Trellet, C. Schmitz, P.L. Kastritis, E. Karaca, A.S.J. Melquiond, M. van Dijk, S.J. de Vries and A.M.J.J. Bonvin. The HADDOCK2.2 webserver: User-friendly integrative modeling of biomolecular complexes. J. Mol. Biol., 428, 720-725 (2015).
-
S.J. de Vries, M. van Dijk and A.M.J.J. Bonvin. The HADDOCK web server for data-driven biomolecular docking. Nature Protocols, 5, 883-897 (2010). Download the final author version here.
Throughout the tutorial, colored text will be used to refer to questions or instructions, and/or PyMOL commands.
This is a question prompt: try answering it! This an instruction prompt: follow it! This is a PyMOL prompt: write this in the PyMOL command line prompt! This is a Linux prompt: insert the commands in the terminal!
Setup/Requirements
In order to follow this tutorial you only need a web browser, a text editor, and PyMOL
(freely available for most operating systems) on your computer in order to visualize the input and output data.
Further, the required data to run this tutorial are the same as for the DisVis tutorial
and should be aquired from this download link.
Once downloaded, make sure to unpack the archive.
HADDOCK general concepts
HADDOCK (see https://www.bonvinlab.org/software/haddock2.4) is a collection of python scripts derived from ARIA (https://aria.pasteur.fr) that harness the power of CNS (Crystallography and NMR System – https://cns-online.org) for structure calculation of molecular complexes. What distinguishes HADDOCK from other docking software is its ability, inherited from CNS, to incorporate experimental data as restraints and use these to guide the docking process alongside traditional energetics and shape complementarity. Moreover, the intimate coupling with CNS endows HADDOCK with the ability to actually produce models of sufficient quality to be archived in the Protein Data Bank.
A central aspect to HADDOCK is the definition of Ambiguous Interaction Restraints or AIRs. These allow the translation of raw data such as NMR chemical shift perturbation or mutagenesis experiments into distance restraints that are incorporated in the energy function used in the calculations. AIRs are defined through a list of residues that fall under two categories: active and passive. Generally, active residues are those of central importance for the interaction, such as residues whose knockouts abolish the interaction or those where the chemical shift perturbation is higher. Throughout the simulation, these active residues are restrained to be part of the interface, if possible, otherwise incurring in a scoring penalty. Passive residues are those that contribute for the interaction, but are deemed of less importance. If such a residue does not belong in the interface there is no scoring penalty. Hence, a careful selection of which residues are active and which are passive is critical for the success of the docking.
The docking protocol of HADDOCK was designed so that the molecules experience varying degrees of flexibility and different chemical environments, and it can be divided in three different stages, each with a defined goal and characteristics:
-
1. Randomization of orientations and rigid-body minimization (it0)
In this initial stage, the interacting partners are treated as rigid bodies, meaning that all geometrical parameters such as bonds lengths, bond angles, and dihedral angles are frozen. The partners are separated in space and rotated randomly about their centers of mass. This is followed by a rigid body energy minimization step, where the partners are allowed to rotate and translate to optimize the interaction. The role of AIRs in this stage is of particular importance. Since they are included in the energy function being minimized, the resulting complexes will be biased towards them. For example, defining a very strict set of AIRs leads to a very narrow sampling of the conformational space, meaning that the generated poses will be very similar. Conversely, very sparse restraints (e.g. the entire surface of a partner) will result in very different solutions, displaying greater variability in the region of binding. -
2. Semi-flexible simulated annealing in torsion angle space (it1)
The second stage of the docking protocol introduces flexibility to the interacting partners through a three-step molecular dynamics-based refinement in order to optimize interface packing. It is worth noting that flexibility in torsion angle space means that bond lengths and angles are still frozen. The interacting partners are first kept rigid and only their orientations are optimized. Flexibility is then introduced in the interface, which is automatically defined based on an analysis of intermolecular contacts within a 5Å cut-off. This allows different binding poses coming from it0 to have different flexible regions defined. Residues belonging to this interface region are then allowed to move their side-chains in a second refinement step. Finally, both backbone and side-chains of the flexible interface are granted freedom. The AIRs again play an important role at this stage since they might drive conformational changes. -
3. Refinement in Cartesian space with explicit solvent (water)
The final stage of the docking protocol immerses the complex in a solvent shell so as to improve the energetics of the interaction. HADDOCK currently supports water (TIP3P model) and DMSO environments. The latter can be used as a membrane mimic. In this short explicit solvent refinement the models are subjected to a short molecular dynamics simulation at 300K, with position restraints on the non-interface heavy atoms. These restraints are later relaxed to allow all side chains to be optimized.
The performance of this protocol of course depends on the number of models generated at each step. Few models are less probable to capture the correct binding pose, while an exaggerated number will become computationally unreasonable. The standard HADDOCK protocol generates 1000 models in the rigid body minimization stage, and then refines the best 200 – regarding the energy function - in both it1 and water. Note, however, that while 1000 models are generated by default in it0, they are the result of five minimization trials and for each of these the 180º symmetrical solution is also sampled. Effectively, the 1000 models written to disk are thus the results of the sampling of 10.000 docking solutions.
The final models are automatically clustered based on a specific similarity measure - either the positional interface ligand RMSD (iL-RMSD) that captures conformational changes about the interface by fitting on the interface of the receptor (the first molecule) and calculating the RMSDs on the interface of the smaller partner, or the fraction of common contacts (FCC), which is the current default, that measures the similarity of the intermolecular contacts. For RMSD clustering, the interface used in the calculation is automatically defined based on an analysis of all intermolecular contacts made in all models.
The information at hand
Let us first inspect the available data, namely the two individual structures (or rather homology models) as well as the information from MS we have at hand to guide the docking.
In the data you downloaded you will find two PDB files for PRE5 (UniProtKB: O14250) and PUP2 (UniProtKB: Q9UT97), the components of the complex we are modeling. If you click on the UniProtKB entries and search for the available structural information, you will see that no experimental structures are available for those. What we will be using here are homology models obtained from SwissModel (this can also be seen in the content of the PDB file if you open it in a text editor for example).
From MS, we have seven experimentally determined cross-links (4 ADH & 3 ZL) (Leitner et al.,
2014).
These are provided in the restraints_filtered.txt file in the data you just downloaded:
# ADH crosslinks A 27 CA B 18 CA 0 23 A 122 CA B 125 CA 0 23 A 122 CA B 128 CA 0 23 A 122 CA B 127 CA 0 23 # ZL crosslinks A 55 CA B 169 CA 0 26 A 55 CA B 179 CA 0 26 A 54 CA B 179 CA 0 26
This is the format used by DisVis to represent the cross-links. Each cross-link definition consists of eight fields:
- chainID of the 1st molecule
- residue number
- atom name
- chainID of the 2nd molecule
- residue number
- atom name
- lower distance limit
- upper distance limit
In addition to those, the DisVis interaction analysis provided us with a list of putative interface residues for each molecule:
PRE5 predicted interface residues: 7,10,13,15,55,58,60,82,83,125,126,127,128,129,131,133 PUP2 predicted interface residues: 1,2,3,5,8,11,13,15,16,17,114,121,122,123,124,140,152,154,177
These correspond to the interface residues identified by DisVis using a cutoff of 0.5 for the average number of interactions as obtained from the following DisVis run.
Note that this cutoff is not a hard limit. However, in the context of using this information to drive the docking in HADDOCK, it is better to be too generous in the definition of the interface rather than too restrictive. Better results are expected if the “true” interface is properly covered. False predictions will not hurt the performance too much since by default HADDOCK will randomly delete for each model a fraction of the provided data.
Visualizing PRE5
Using PyMOL, let us first take a look at PRE5 and visualize the information from MS.
For this open the PDB file PRE5.pdb.
PyMOL Menu → File → Open… → Select the file
If you want to use the PyMOL command-line instead, type the following command:
As a first step, we will color in orange all residues identified by the DisVis interaction analysis. Enter following commands at the Command Line in PyMOL:
And then we will color in red all residues involved in a cross-link:
Do the MS-derived residues suggest a well-defined interface?
You can also switch to a surface representation:
See solution:
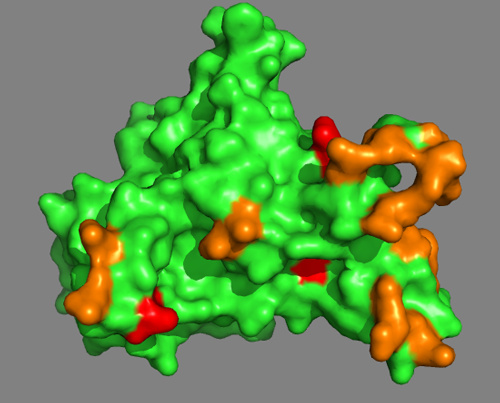
Visualizing PUP2
Using PyMOL, let us now take a look at PUP2 and visualize the information from MS.
For this open the PDB file PUP2.pdb.
PyMOL Menu → File → Open… → Select the file
If you want to use the PyMOL command-line instead, type the following command:
As a first step, we will color in orange all residues identified by the DisVis interaction analysis. Enter following commands at the Command Line in PyMOL:
And then we will color in red all residues involved in a cross-link:
color red, resid 18+125+127+128+169+179
Do the MS-derived residues suggest a well-defined interface?
You can also switch to a surface representation:
See solution:
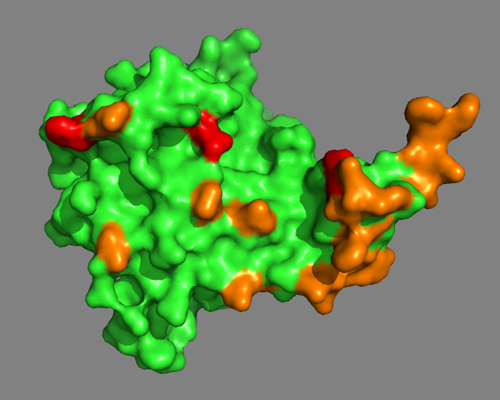
Scenarios for docking - how to make use of MS data in HADDOCK
We have two types of data:
- The
cross-links detected by MSthat provide distance restraints between pairs of residues/atoms - The
derived information about putative interface residuesobtained from the DisVis interaction analysis
In this tutorial we will investigate three different scenarios to guide the docking in HADDOCK:
-
Using the cross-links as distance restraints, supplemented by center-of-mass restraints in HADDOCK. The latter option is added to ensure more compact solutions since the distances defined by the cross-links are rather loose (this is defined by the chemistry and flexibility of the molecules used for the cross-link reaction). -
Using the derived interface residues to define ambiguous interaction restraints. Those residues will be input as active residues in HADDOCK. These do not define any specific pairwise distance, but rather a network of ambiguous distances that will pull the interface together without pre-defining their orientation. -
A combination of cross-link restraints and derived interface information(i.e. the combination of options 1 and 2).
For setting up the docking runs we will be using the expert interface of the HADDOCK server for scenarios 1 and 3 and the easy interface for scenario 2.
Before setting up the docking we need first to generate the distance restraint file for the cross-links in a format suitable for HADDOCK. HADDOCK uses CNS as computational engine. A description of the format for the various restraint types supported by HADDOCK can be found in our Nature Protocol paper, Box 4.
Distance restraints are defined as:
assign (selection1) (selection2) distance, lower-bound correction, upper-bound correction
The lower limit for the distance is calculated as: distance minus lower-bound correction and the upper limit as: distance plus upper-bound correction
The syntax for the selections can combine information about chainID - segid keyword -, residue number - resid
keyword -, atom name - name keyword.
Other keywords can be used in various combinations of OR and AND statements. Please refer for that to the online CNS manual.
Here would be an example of a distance restraint between the CB carbons of residues 10 and 200 in chains A and B with an allowed distance range between 10 and 20Å:
assign (segid A and resid 10 and name CB) (segid B and resid 200 and name CB) 20.0 10.0 0.0
See solution:
assign (segid A and resid 27 and name CA) (segid B and resid 18 and name CA) 23 23 0 assign (segid A and resid 122 and name CA) (segid B and resid 125 and name CA) 23 23 0 assign (segid A and resid 122 and name CA) (segid B and resid 128 and name CA) 23 23 0 assign (segid A and resid 122 and name CA) (segid B and resid 127 and name CA) 23 23 0 assign (segid A and resid 55 and name CA) (segid B and resid 169 and name CA) 26 26 0 assign (segid A and resid 55 and name CA) (segid B and resid 179 and name CA) 26 26 0 assign (segid A and resid 54 and name CA) (segid B and resid 179 and name CA) 26 26 0
Note: Under Linux (or OSX), this file could be generated automatically from the restraints_filtered.txt
file provided with the data for this tutorial by giving the following command (one line) in a terminal window:_
Register for the HADDOCK web server
Register for getting access to the web server (or use the credentials provided to you in case of a workshop).
Fill the required information. Registration is not automatic but is usually processed within 12h, so be patient. Once you got your credentials, you will need to request access to the expert server in order to complete this tutorial.
Setting up the docking from cross-link restraints (scenario 1)
We will now launch the docking run. For this scenario we will make us of the expert interface of the HADDOCK web server:
https://wenmr.science.uu.nl/services/HADDOCK2.2/haddockserver-expert.html
Note: The blue bars on the server can be folded/unfolded by clicking on the arrow on the right.
-
Step1: Define a name for your docking run, e.g. PRE5-PUP2-MS-crosslinks.
-
Step2: Input the first protein PDB file. For this unfold the First Molecule menu.
First molecule: where is the structure provided? -> “I am submitting it” Which chain to be used? -> All (for this particular case) PDB structure to submit -> Browse and select PRE5.pdb Segment ID to use during docking -> A
- Step3: Input the second protein PDB files. For this unfold the Second Molecule menu.
Second molecule: where is the structure provided? -> “I am submitting it” Which chain to be used? -> All (for this particular case) PDB structure to submit -> Browse and select PUP2.pdb Segment ID to use during docking -> B
- Step4: Input the cross-links distance restraints and turn on center-of-mass restraints. For this unfold the Distance Restraint menu.
Input under the unambiguous distance restraints the cross-links distance restraints file you created Center of mass restraints -> Check the box
- Step 5: You are ready to submit!
Enter your username and password (or the course credentials provided to you).
If the input fields have been correctly filled you should be redirected to a status page displaying a message indicating that your run has been successfully submitted. You will first be presented with a web page containing a link to the results page, but also an important link to a haddockparameter file (simple text format) containing all settings and input data of your run.
We strongly recommend to save this haddockparameter file since it will allow you to repeat the run by simply uploading it into the file upload interface of the HADDOCK webserver. It can thus serve as input reference for the run. This file can also be edited to change a few parameters, for example increasing the number of models generated.
An excerpt of this file is shown here:
HaddockRunParameters ( runname = 'PRE5-PUP2-MS-crosslinks', auto_passive_radius = 6.5, create_narestraints = True, delenph = True, ranair = False, cmrest = True, ...
This file contains all parameters and input data of your run, including the uploaded PDB files and the restraints.
Can you locate the distance restraints in this file?
Click now on the link to the results page. While your input data are being validated and processed the page will show:

During this stage the PDB and eventually provided restraint files are being validated. Further the server makes use of Molprobity to check side-chain conformations, eventually swap them (e.g. for asparagines) and define the protonation state of histidine residues. Once this has been successfully done, the page will indicate that your job is first QUEUED, and then RUNNING.

The page will automatically refresh and the results will appear upon completion (which can take between 1/2 hour to several hours depending on the size of your system and the load of the server). You will be notified by email once your job has successfully completed.
Setting up the docking with the DisVis-derived interfaces (scenario 2)
For this scenario we will make us of the easy interface of the HADDOCK web server:
https://wenmr.science.uu.nl/services/HADDOCK2.2/haddockserver-easy.html
-
Step1: Define a name for your docking run, e.g. PRE5-PUP2-MS-interface.
-
Step2: Input the first protein PDB file. For this unfold the First Molecule menu.
First molecule: where is the structure provided? -> “I am submitting it” Which chain to be used? -> All (for this particular case) PDB structure to submit -> Browse and select PRE5.pdb Active residues (directly involved in the interaction) -> 7,10,13,15,55,58,60,82,83,125,126,127,128,129,131,133 Define passive residues automatically around the active residues -> check box
- Step3: Input the second protein PDB files. For this unfold the Second Molecule menu.
Second molecule: where is the structure provided? -> “I am submitting it” Which chain to be used? -> All (for this particular case) PDB structure to submit -> Browse and select PUP2.pdb Active residues (directly involved in the interaction) -> 1,2,3,5,8,11,13,15,16,17,114,121,122,123,124,140,152,154,177 Define passive residues automatically around the active residues -> check box
- Step 4: You are ready to submit!
Enter your username and password (or the course credentials provided to you).
Remember to save the haddockparameter file for reference.
Setting up the docking with cross-links and the DisVis-derived interfaces (scenario 3)
For this scenario we will make us of the expert interface of the HADDOCK web server:
https://alcazar.science.uu.nl/services/HADDOCK2.2/haddockserver-expert.html
-
Step1: Define a name for your docking run, e.g. PRE5-PUP2-MS-crosslinks-interface.
-
Step2: Input the first protein PDB file. For this unfold the First Molecule menu.
First molecule: where is the structure provided? -> “I am submitting it” Which chain to be used? -> All (for this particular case) PDB structure to submit -> Browse and select PRE5.pdb Active residues (directly involved in the interaction) -> 7,10,13,15,55,58,60,82,83,125,126,127,128,129,131,133 Define passive residues automatically around the active residues -> check box
- Step3: Input the second protein PDB files. For this unfold the Second Molecule menu.
Second molecule: where is the structure provided? -> “I am submitting it” Which chain to be used? -> All (for this particular case) PDB structure to submit -> Browse and select PUP2.pdb Active residues (directly involved in the interaction) -> 1,2,3,5,8,11,13,15,16,17,114,121,122,123,124,140,152,154,177 Define passive residues automatically around the active residues -> check box
- Step4: Input the cross-links distance restraints and turn on center-of-mass restraints. For this unfold the Distance Restraint menu.
Input under the unambiguous distance restraints the cross-links distance restraints file you created Center of mass restraints -> Check the box
- Step 5: You are ready to submit!
Enter your username and password (or the course credentials provided to you).
Remember to save the haddockparameter file for reference.
First analysis of the results
Once your run has completed you will be presented with a result page showing the cluster statistics and some graphical representation of the data (and if registered, you will also be notified by email). If using course credentials provided to you, the number of models generated will have been decreased to allow the runs to complete within a reasonable amount of time. Because of that, the results might not be very good.
We already pre-calculated full docking runs (meaning that the default number of models has been generated: 1000 for rigid-body docking and 200 for semi-flexible and the final refinement). The full runs for the three scenarios described above can be accessed at:
-
Scenario 1: https://alcazar.science.uu.nl/services/HADDOCK2.2/Files/PRE5-PUP2-MS-crosslinks/
-
Scenario 2: https://alcazar.science.uu.nl/services/HADDOCK2.2/Files/PRE5-PUP2-MS-interface/
-
Scenario 3: https://alcazar.science.uu.nl/services/HADDOCK2.2/Files/PRE5-PUP2-MS-crosslinks-interface/
Example result page
For each of the three scenarios:
Inspect the result page How many clusters are generated?
Note: The bottom of the page gives you some graphical representations of the results, showing the distribution of the solutions for various measures (HADDOCK score, van der Waals energy, …) as a function of the RMSD from the best generated model (the best scoring model).
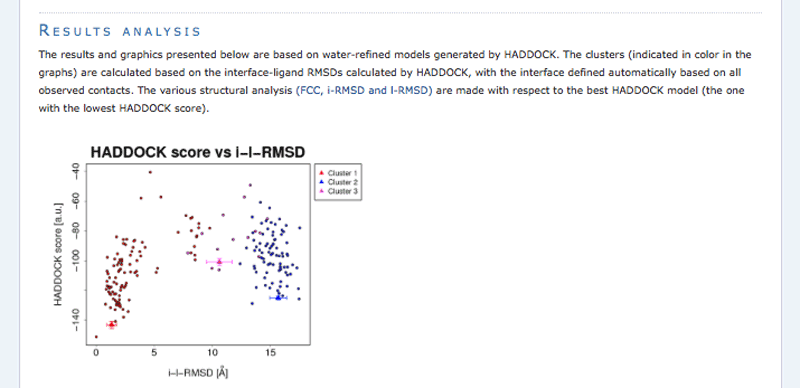
The ranking of the clusters is based on the average score of the top 4 members of each cluster. The score is calculated as:
HADDOCKscore = 1.0 * Evdw + 0.2 * Eelec + 1.0 * Edesol + 0.1 * Eair
where Evdw is the intermolecular van der Waals energy, Eelec the intermolecular electrostatic energy, Edesol represents an empirical desolvation energy term adapted from Fernandez-Recio et al. J. Mol. Biol. 2004, and Eair the AIR energy. The cluster numbering reflects the size of the cluster, with cluster 1 being the most populated cluster. The various components of the HADDOCK score are also reported for each cluster on the results web page.
Consider the cluster scores and their standard deviations. Is the top ranked cluster significantly better than the second one? (This is also reflected in the z-score).
In case the scores of various clusters are within the standard deviation from each other, all should be considered as a valid solution for the docking. Ideally, some additional independent experimental information should be available to decide on the best solution.
Visualisation of docked models
Let’s now visualize the various solutions for the three different scenarios
We illustrate the procedure using the results from scenario 1. You can repeat it for the other scenarios. Start PyMOL and load each cluster representative:
File menu -> Open -> select cluster1_1.pdb
Repeat this for each cluster. Once all files have been loaded, type in the PyMOL command window:
show cartoon
util.cbc
hide lines
Let’s then superimpose all models on chain A of the first cluster:
select cluster1_1 and chain A
align cluster2_1, sele
Repeat the align command for each cluster representative.
This will align all clusters on chain A (PRE5), maximizing the differences in the orientation of chain B (PUP2).
Examine the various clusters. How does the orientation of PUP2 differ between them?
Note: You can turn on and off a cluster by clicking on its name in the right panel of the PyMOL window.
Let’s now check if the solutions actually fit the cross-links we defined.
Start a new PyMOL session and load as described above the model you want to analyze, e.g. the best model of the top
ranking cluster cluster1_1.pdb for scenario 1.
In the PyMOL command window type:
This will draw lines between the connected atoms and display the corresponding Euclidian distance. Objects are created in the left panel with their name corresponding to the cross-link and its associated maximum distance.
See solution:
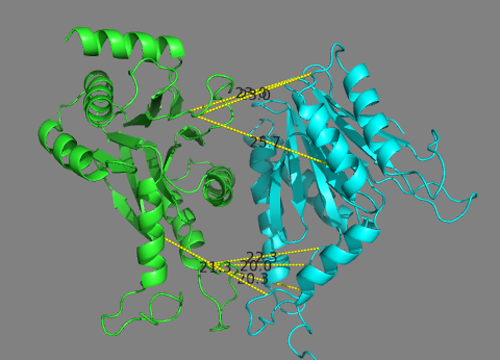
Inspect the various cross-link distances.
Is the model satisfying the cross-link restraints?
Note _that the reported distances are Euclidian distances. In reality, the cross-linker will have to follow the surface of the molecule which might results in a longer effective distance. A proper comparison would required calculating the surface distance instead. Such an analysis can be done with the XWalk sofware.
We can also visualize the interface residues identified from the DisVis interaction analysis. For this type in the PyMOL command window:
For better visualization turn on the surface representation:
Are all predicted interface residues from DisVis forming contacts?
See solution:
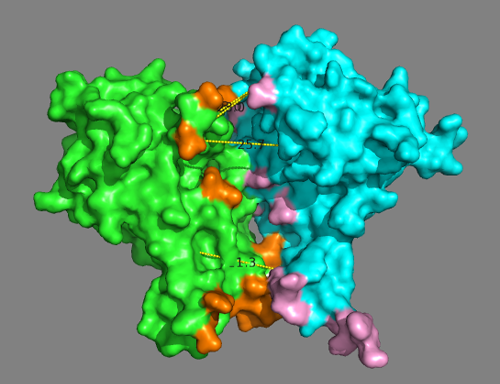
While the model is mostly satisfying the defined distance restraints, many residues identified as potential interface by DisVis are not forming contacts.
Now repeat this analysis for the top ranking model of scenario 2 and 3.
Is the model satisfying the cross-link restraints? Are all predicted interface residues from DisVis forming contacts?
See solution for scenario 2:
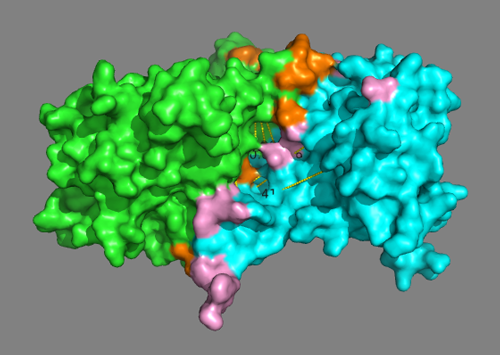
In this model some cross-links are severely violated with distances > 40Å. But the putative interface residues are mostly forming contacts.
See solution for scenario 3:
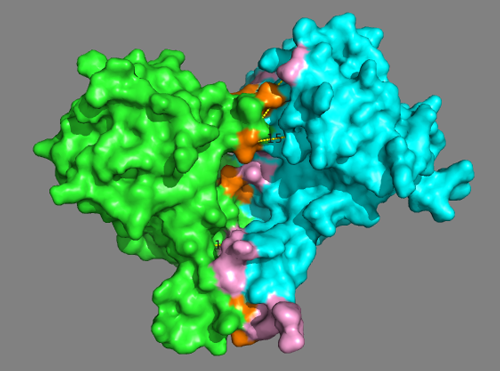
This model nicely satisfies both the cross-links and the predicted interface residues from the DisVis interaction analysis.
Comparison with the crystal structure of an homologous complex
In order to check if any of the docking models we obtained using the various scenarios makes sense,
we will compare them to a recently published article (November 2016) structure of a homologue
of the 26S proteasome (from S. cerevisiae). This structure has been solved by X-ray crystallography at 2.4Å resolution (PDBid
5L5A).
We will only use the two chains that are of interest, namely chains D and C corresponding to PRE5 and PUP2 respectively.
The corresponding PDB file is available in the downloaded tutorial data as 5l5a_CD.pdb.
For each of the three docking scenarios we will visualize the top ranking model of each cluster and compare those to the crystal structure of the homologous complex. We will only illustrate this process for scenario 3, for which the top 10 clusters correspond to 1-9 and 11.
Open PyMOL and load the top ranking model of each cluster
Also load the structure of the homologous complex -> 5l5a_CD.pdb
In PyMOL type the following commands (the last two lines are for removing water from the crystal structure):
show cartoon
util.cbc
hide lines
select 5l5a_CD and resn HOH
remove sele
Let’s now superimpose all models onto the crystal structure, using PRE5 to emphasize the difference in the orientation of PUP2. For this we need to superimpose chain A of our models onto chain D of the homologous crystal structure.
You now see all models superimposed on the the crystal structure of the homologous complex (shown in yellow and magenta). Turn on and off models (by clicking on the name in the right panel).
Can you identify any cluster that resembles the crystal structure?
If this is the case, what is the rank of that cluster in the HADDOCK ranking?
Note that the cluster number does not indicate its rank. The clusters are listed in the HADDOCK result page in the order of their ranking. The cluster number only reflects the size of the cluster, with cluster1 being the most populated.
See solution for scenario 3:
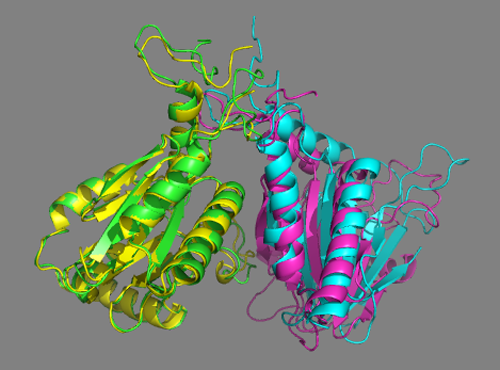
Cluster1 of the scenario 3 docking run using both cross-links and the interfaces predicted by DisVis nicely matches the crystal structure. This is also the top-ranking cluster according to HADDOCK. </a>
Now repeat this analysis, but for the other two scenarios
Does the docking using only the cross-links as distance restraints generate any reasonable models?
See solution for scenario 1:
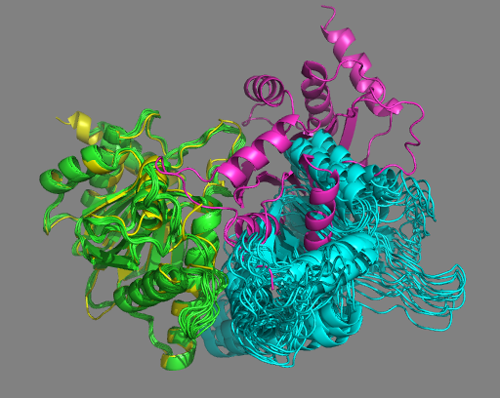
No single cluster matches the crystal structure in this case.
See solution for scenario 2:
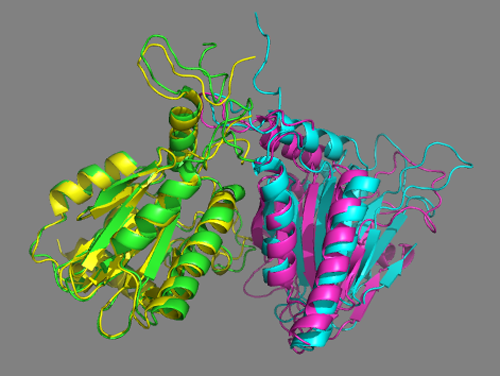
Cluster9 of the scenario 2 docking run using the interfaces predicted by DisVis nicely matches the crystal structure. Cluster9 in this case is the second-ranking cluster. Its score (-85+/-12) is higher (the lower the score the better) than the top-ranking cluster (-104+/-21), but both cluster have large standard deviations in their score and overlap. </a>
Conclusions
We have demonstrated the use of cross-linking data from mass spectrometry for guiding the docking process in HADDOCK. Because of the rather low precision of the detected cross-links (meaning here their rather large distance range), using them alone to guide docking in HADDOCK might not be the best scenario.
Additional information on the putative interfaces can be extracted by performing an interaction analysis in our DisVis web portal. By performing an exhaustive search of all geometrically feasible complexes consistent with the cross-linking data, DisVis can extract key interacting residues. This is additional information that might be beneficial to guide the docking as demonstrated here.
We should also note that the modeling was done from models of PRE5 and PUP2, with their limitations. If better, higher accuracy structures are available the results might well be different. Still, from this analysis, the recommended scenario for docking using our HADDOCK software would be the combination of distance restraints representing the cross-links with interface information extracted from the DisVis interaction analysis.
Congratulations!
Thank you for following this tutorial. If you have any questions or suggestions, feel free to contact us via email, or post your question to
our HADDOCK forum hosted by the
![]() Center of Excellence for Computational Biomolecular Research.
Center of Excellence for Computational Biomolecular Research.