Welcome to the haddock-restraints guide
haddock-restraints is a command-line tool that helps you to generate restraints for the HADDOCK docking software.
The generation of restraints is a crucial step in the preparation of a docking experiment. The restraints define the spatial constraints that guide the docking software in the search for the best conformation of the complex.
HADDOCK uses a .tbl format to define the restraints. If you are using the web service, these are defined automatically via the web interface. However, if you are running HADDOCK locally, you need to provide these restraints yourself.
This is where haddock-restraints comes in. It helps you to generate the restraints in the .tbl format, based on the input structures you provide.
See the INSTALLATION section to get started and proceed to the USAGE section to learn how to use the tool.
Getting help
If you encounter any issues or have any questions, please open an issue on the GitHub repository, contact us at bonvinlab.support@uu.nl or join the BioExcel forum and post your question there.
Installation
haddock-restraints is a Rust command-line tool. You can either download the pre-compiled binary, download from the Rust package manager or build it from source.
Download
Get a pre-compiled binary for your system at the link below:
Build from Cargo
To install the tool using Cargo, you need to have the Rust toolchain installed on your system. You can install Rust by following the instructions on the official website.
cargo install haddock-restraints
Now it should be available in your system. You can check if it is installed by running:
haddock-restraints -h
From source
If you prefer to install it from source, you can clone the repository and build it using Cargo:
git clone https://github.com/haddocking/haddock-restraints
cd haddock-restraints
cargo build --release
Go ahead and proceed to the USAGE section to learn how to use the tool.
Usage
haddock-restraints is a command-line tool that is organized with subcommands.
Each subcommand has its own set of options and arguments. The main subcommands are:
-
restraint: Generate unambiguous restraints to keep molecules together during docking -
z: Generate restraints to keep the molecule aligned to the Z-axis
Generate .tbl file from a configuration file
haddock-restraints can generate a .tbl file from a configuration file. The configuration file is a JSON file that contains the information needed to generate the restraints.
To use this file you first need to get familiar with a few concepts:
Usage
To run the tbl subcommand, you just need to provide the path to the configuration file. For example:
haddock-restraints tbl path/to/config.json > restraints.tbl
Active/Passive residues
We have a deeper explanation of these at our Best Practice Guide - but for brevity , we can say that:
- Active residues MUST be in contact with the other molecule.
- Passive residues COULD be in contact with the other molecule.
Let's say you have two proteins A and B; for protein A, you have very high confidence, based on your experiments, in information retrieved from the literature or bioinformatic predictions that residue number 950 is very important for the interaction.
However for protein B you do not know the exact residues involved in the interaction, but you know that the interaction happens in a specific region of the protein. Let's say this region is between residues 41 and 45.
In this example, your active residues are:
- Protein A: 950
- Protein B: 41, 42, 43, 44, 45
For your passive residues, you can select a region around your active residues to serve as the passive residues. This is a way to tell the docking software that these residues are not as important as the active ones, but they could be in contact with the other molecule.
You can do the passive selection manually, by visual inspection or
haddock-restraintscan do it automatically for you, using thepassive_from_activeoption.
In the end, your configuration file will look like this:
[
{
"id": 1,
"chain": "A",
"active": [950],
"passive": [],
"structure": "proteinA.pdb",
"passive_from_active": true,
"target": [2]
},
{
"id": 2,
"chain": "B",
"active": [41, 42, 43, 44, 45],
"passive": [],
"structure": "proteinB.pdb",
"passive_from_active": true,
"target": [1]
}
]
Visually, the restraints generated by this configuration file will look like this:
| Protein A | Protein B |
|---|---|
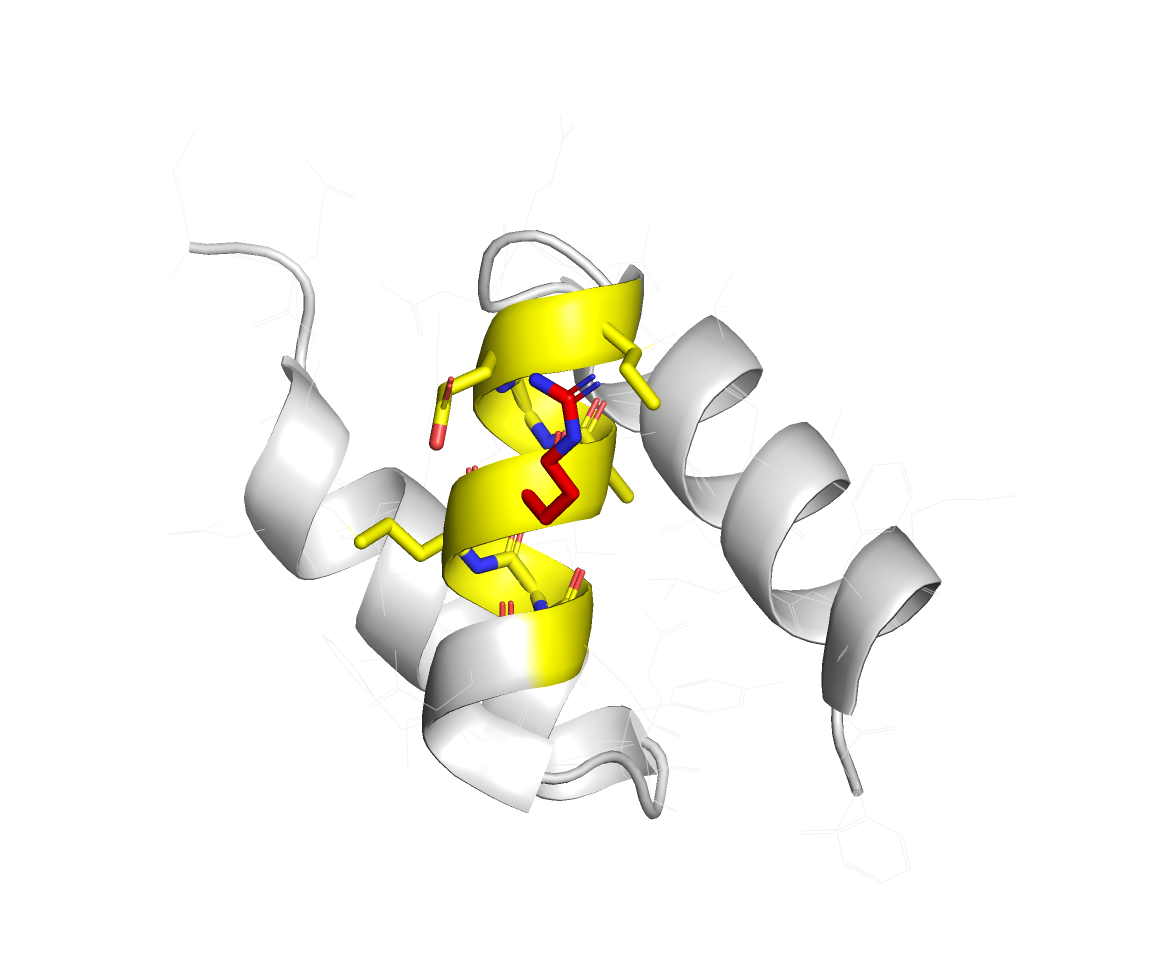 | 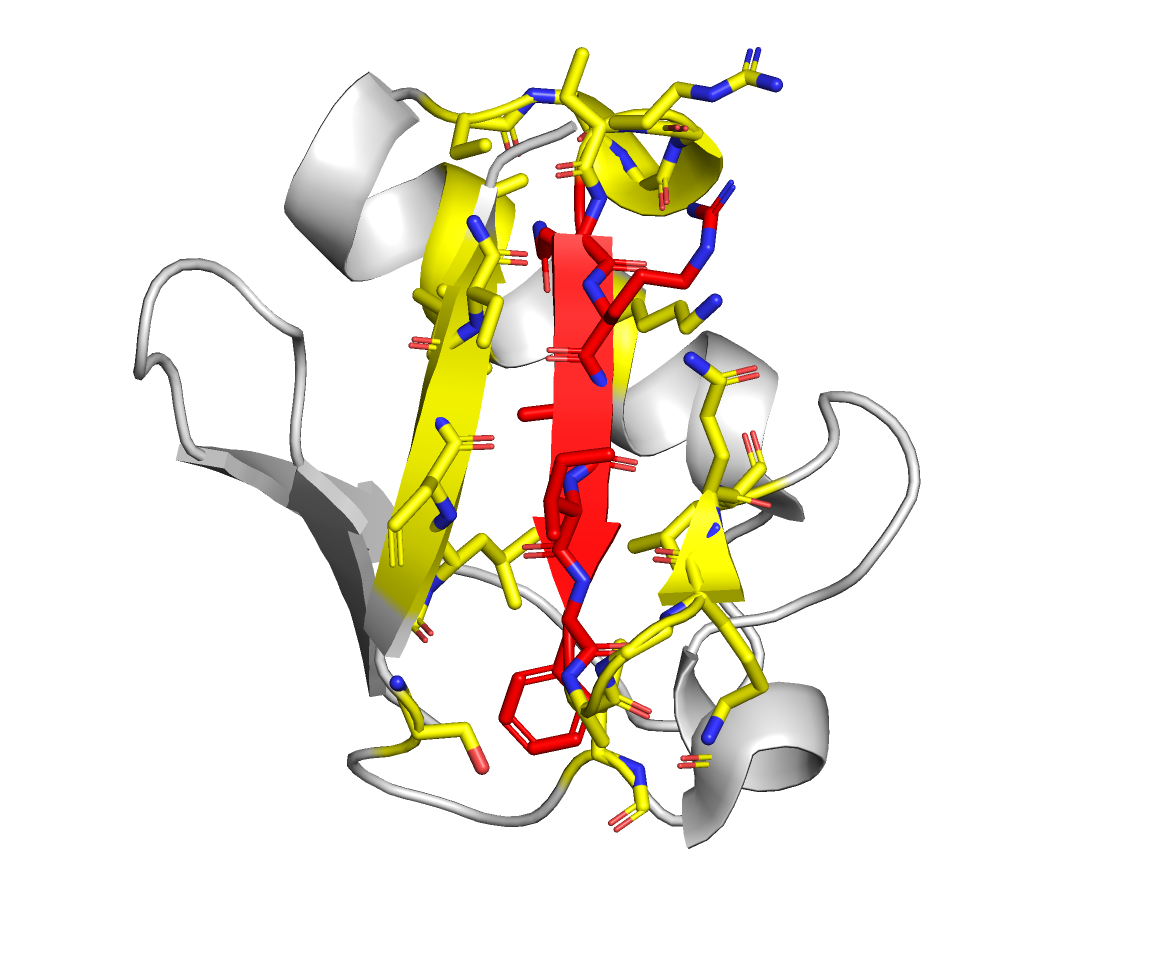 |
Active residues are in red, and the passive residues are in yellow.
tbl configuration file
Restraints are very powerful tools to guide the docking process, to use its full potential, in haddock-restraints we use a configuration file to define the restraints. This file is a JSON file that contains the information needed to generate the restraints.
To use the full potential of ambiguous restraints we have introduced the concept of interactors, which are arbitrary groups of residues that can be used to define the restraints.
haddock-restraints supports an unlimited number of interactors, and each interactor can have an unlimited number of residues. This allows you to define complex restraints that can be used to guide the docking process.
- Mandatory fields
- Optional fields
- Reference guide with detailed explanation of the fields in the configuration file
Mandatory fields
id: an integer that identifies the interactorchain: the chain of the interactoractive: a list of residues that are active in the interactionpassive: a list of residues that are passive in the interactiontarget: a list of integers that identifies the interactors that the current interactor interacts with
A minimal configuration file would look like this:
[
{
"id": 1,
"chain": "A",
"active": [950],
"passive": [],
"target": [2]
},
{
"id": 2,
"chain": "B",
"active": [41, 42, 43, 44, 45],
"passive": [],
"target": [1]
}
]
Optional fields
structure: the PDB file that contains the structure of the interactorpassive_from_active: if true, the passive residues are defined based on the active residues (requires structure)passive_from_active_cutoff: radius used to search for passive residues around the active (requires structure)surface_as_passive: if true, the passive residues are defined based on the surface accessibility of the residues (requires structure)filter_buried: if true, the buried residues are filtered out (requires structure)filter_buried_cutoff: the cutoff to consider a residue as buried, default = 0.7 (requires structure)target_distance: the distance to consider two residues as interacting, default = 2.0 (see Note)lower_margin: the lower bound correction subtracted from thetarget_distanceto define the lower distance boundary.upper_margin: the upper bound correction added to the target_distance to define the upper distance boundary.active_atoms: a list containing which active atoms should be included in the restraint; if not defined all atoms are included automaticallypassive_atoms: a list containing which passive atoms should be included in the restraint; if not defined all atoms are included automatically
Note: Check this paper for a deeper explanation about the target distance and the margins. The default value of 2.0Å (which might seem short) is used because of the way the effective distance is calculated from all pairwise combinations of atom it will always be shorter than the shortest distance measured.
A configuration file with optional fields would look like this:
[
{
"id": 1,
"chain": "A",
"active": [934, 939],
"passive": [],
"structure": "2oob.pdb",
"target": [2],
"passive_from_active": true,
"filter_buried": true
},
{
"id": 2,
"chain": "B",
"active": [68],
"passive": [],
"target": [1]
},
{
"id": 3,
"chain": "B",
"active": [],
"passive": [],
"target": [1],
"structure": "2oob.pdb",
"surface_as_passive": true
}
]
Reference guide
-
id- type: integer
- description: an integer that identifies the interactor
-
chain- type: string
- description: the chain of the interactor
-
active- type: list of integers
- description: a list of residues that are active in the interaction
-
passive- type: list of integers
- description: a list of residues that are passive in the interaction
-
target- type: list of integers
- description: a list of integers that identifies the interactors that the current interactor interacts with
Optional fields are:
-
structure- type: string
- description: the PDB file that contains the structure of the interactor. If using relative paths, they should be relative to the configuration file
-
passive_from_active- type: boolean
- description: define passive residues are defined based on the active residues
- requires:
structure
-
surface_as_passive- type: boolean
- description: define passive residues are defined based on the surface accessibility of the residues
- requires:
structure
-
filter_buried- type: boolean
- description: filter out buried residues
- requires:
structure
-
filter_buried_cutoff- type: float
- description: the cutoff to consider a residue as buried, default = 0.7
- requires:
structure
-
target_distance- type: float
- description: the distance to consider two residues as interacting, default = 2.0
-
lower_margin- type: float
- description: the lower bound correction subtracted from the
target_distanceto define the lower distance boundary
-
upper_margin- type: float
- description: the upper bound correction added to the target_distance to define the upper distance boundary
Generate true-interface restraints from a PDB file
This is a very specific type of restraint, that is used to restrain the interface of a protein-protein complex most commonly used to benchmark the efficiency of a docking workflow or protocol.
What this command does is to calculate the distance between the residues of two chains and if the distance is less than a given cutoff, the residues are considered to be in the interface.
Based on this, haddock-restraints fills in the active and passive fields and provides you with a .tbl file that can be used to restrain the interface of the protein-protein complex.
Usage
To run the ti subcommand, you just need to provide the path to the PDB file and the cutoff distance. For example:
haddock-restraints ti path/to/complex.pdb 5.0 > ti.tbl
Generate unambiguous restraints to keep molecules together during docking
In some scenarios, you might be docking proteins that contain
structural gaps, but you still want to keep the molecules and ligands
in their place together during the docking process. This is where the
restraint subcommand comes in handy.
It will generate unambiguous restraints to keep the molecules together during docking.
Usage
To run the restraint subcommand, you just need to provide the path to
the PDB file. For example:
haddock-restraints restraint path/to/complex.pdb > unambig.tbl
List residues in the interface
This command is a helpful when you want to know which residues are in the interface of a protein-protein complex. It calculates the distance between the residues of two chains and if the distance is less than a given cutoff, the residues are considered to be in the interface.
The result of this might be useful to generate restraints using the
tblcommand - to define active and/or passive residues.
Usage
To run the interface subcommand, you just need to provide the path to the PDB file and the cutoff distance. For example:
haddock-restraints interface path/to/complex.pdb 5.0
Generate z restraints to keep the molecule aligned to the Z-axis
This subcommand generates restraints to keep the molecule aligned to the Z-axis. This is useful when you want to keep the molecule in a specific orientation during docking.
As input you need to pass at least one selection of residues, this will be used to define a plane perpendicular to the Z-axis. The restraints will be generated to keep the molecule aligned to this plane.
Usage
To run the z subcommand, you need to provide the path to the PDB file, the selection of residues, the output file which will contain the shape beads, the number of beads to generate, and the distance between the beads. For example:
./haddock-restraints z \
--residues 19,83,145,119,167 \
path/to/the/input.pdb \
path/to/the/output/shape.pdb \
20 \ # spacing between the beads in angstrom - 20A
6 \ # grid size in dimension - 6x6
> z_restraints.tbl
Further, follow the HADDOCK documentation <pending> to use the generated restraints in the docking protocol.
Generate unambiguous true-interface restraints from a PDB file
This is a more strict version of the true-interface restraints that assigns specific pairs to each residue in the interface.
For each atom in each of the chains, the algorithm defines the pairs based on atomic interactions between any atom of a given residue in the interface and any other atom in an interacting chain. The restraints are later defined considering the CA-CA distances of such pairs; thus allowing for side-chain flexibility.
Usage
To run the unambig-ti subcommand, you just need to provide the path to the
PDB file and the cutoff distance. For example:
haddock-restraints unambig-ti path/to/complex.pdb 5.0 > unambig.tbl
Development
haddock-restraints is an open-source project. You can contribute to the project by submitting issues, feature requests, or pull requests.
Please go to the GitHub repository to open an issue or submit a pull request.